generalidade
Craniossinostose é o termo pelo qual os médicos indicam uma anormalidade do crânio devido à fusão prematura de uma ou mais suturas cranianas.
As suturas cranianas são as articulações fibrosas que unem os ossos da abóbada craniana (isto é, os ossos frontal, temporal, parietal e occipital).
A craniossinostose pode ser um fenômeno isolado (craniossinostose não sindrômica) ou o resultado de algumas condições mórbidas específicas (craniossinostose sindrômica). Entre as condições mórbidas que causam a fusão prematura das suturas cranianas, as mais conhecidas são: síndrome de Crouzon e síndrome de Apert.
Com a fusão prematura das suturas cranianas, as estruturas encefálicas não possuem o espaço adequado para o crescimento. Isso tem várias conseqüências, incluindo principalmente o aumento da pressão intracraniana (hipertensão intracraniana).
Um diagnóstico oportuno e preciso permite planejar um tratamento ad hoc . Este último é do tipo cirúrgico e tem como objetivo final a separação das suturas fusiformes precocemente.
Recordações da anatomia do crânio humano
Fornecido com ossos e cartilagens, o crânio é a estrutura esquelética da cabeça que constitui a face e protege o cérebro, o cerebelo, o tronco cerebral e os órgãos sensoriais.

Para simplificar o estudo e a compreensão do crânio, os anatomistas pensaram em dividi-lo em dois compartimentos, chamados neurocranium e splancnocranium .
neurocranium
O neurocrânio é a região craniana superior que contém o cérebro e alguns dos principais órgãos sensoriais. Seus ossos mais importantes - estritamente planos - são os ossos frontal, temporal, parietal e occipital; estes, juntos, formam a chamada abóbada craniana .
splanchnocranium
O splanchnocranium, ou facial massivo, é a região ântero-inferior do crânio, composta de ossos pares e desiguais. Representa a estrutura esquelética da face, por isso contém elementos ósseos como a mandíbula, a mandíbula superior, as maçãs do rosto, o osso nasal etc.
O que é craniossinostose?
A craniossinostose é uma anomalia rara do crânio, caracterizada por um formato de cabeça não natural devido à fusão prematura de uma ou mais suturas cranianas . As suturas cranianas são as articulações fibrosas que unem os ossos da abóbada craniana (isto é, os ossos frontal, temporal, parietal e occipital).

Do site: //www.wkomsi.com/
QUANDO DEVE FECHAR O ENCERRAMENTO DAS SUTURAS CRANIANAS?
Em condições normais, a fusão das suturas cranianas ocorre no período pós-natal (NB: alguns processos terminam até aos 20 anos). Este longo processo de fusão permite que o cérebro cresça e se desenvolva adequadamente.
Se, como no caso da craniossinostose, a fusão ocorrer cedo demais - portanto durante a vida pré-natal, perinatal ou infantil - os elementos encefálicos (cérebro, cerebelo e tronco cerebral) e alguns órgãos dos sentidos (olhos em particular) uma alteração de forma e crescimento.
causas
O processo fisiopatológico que determina a craniossinostose é a fusão prematura das suturas cranianas .
Esse processo pode representar um fenômeno isolado - onde "isolado" significa que não está associado a nenhum estado mórbido em particular - ou pode ser a consequência de algumas síndromes específicas, quase sempre de natureza genética.
Em vista disso, os médicos pensaram em classificar a craniossinostose em duas categorias:
- Craniosinostose não sindrômica . O termo não-sindrômico indica que a anomalia craniana não está associada a nenhuma patologia ou outro defeito físico.
- A craniossinostose sindrômica . O termo sindrômico significa que a malformação craniana é o resultado de uma síndrome particular, na maioria dos casos do tipo genético.
CRANIOSINOSTOSE NÃO SINDRÔMICA
Médicos e pesquisadores ainda não estabeleceram as causas da craniossinostose não sindrômica.
Eles propuseram várias hipóteses - incluindo a influência de fatores ambientais ou problemas semelhantes a hormônios - mas nenhuma dessas teorias foi confirmada por resultados experimentais.
Portanto, para entender a origem precisa da anomalia, mais estudos são necessários.
CRANIOSINOSTOSE SINDROMICA
Segundo as últimas pesquisas médicas, existem mais de 150 síndromes diferentes, todas muito raras, capazes de causar craniossinostose.
Entre essas síndromes, as mais conhecidas e comuns são:
- Síndrome de Crouzon . O resultado de mutações específicas nos genes FGFR2 (cromossomo 10) e FGFR3 (cromossomo 4), essa condição mórbida afeta um recém-nascido a cada 60.000 e envolve a presença de anormalidades exclusivamente no nível da cabeça e da face.
- Síndroma de Apert Ocorre principalmente devido a mutações no gene FGFR2 (o mesmo que a síndrome de Crouzon) e afeta um recém-nascido a cada 100.000 ou mais.
Diferentemente da síndrome de Crouzon, as alterações genéticas do FGFR2 são tais que as malformações envolvem não apenas o crânio e a face, mas também as mãos e os pés.
- Síndrome de Pfeiffer . Surge devido a mutações no gene FGFR2 "usual" e um gene com funções similares, chamado FGFR1 (cromossomo 8). A peculiaridade dessas mutações é que - além das deformidades do crânio e da face - elas também determinam: sindactilia, braquidactilia e polegares e dedos grandes (desproporcionais aos outros dedos).
A síndrome de Pfeiffer tem uma incidência de um em cada 100.000 recém-nascidos.
- Síndrome de Saethre-Chotzen . É uma condição genética que afeta um recém-nascido a cada 50.000 ou mais. Provoca várias malformações ao nível do crânio, face, mãos e pés. Algumas mutações específicas do gene TWIST1, localizadas no cromossomo 7, são responsáveis pelo surgimento da síndrome de Saethre-Chotzen.
EPIDEMIOLOGIA DA CRANIOSINOSTOSE
De acordo com as estatísticas mais recentes, parece que uma criança de cerca de 1800-3000 sofre de craniossinostose.
Quanto ao sexo mais afetado, vários estudos clínicos mostraram que 3 de 4 pacientes são do sexo masculino. A razão pela qual a craniossinostose é mais prevalente na população masculina é completamente desconhecida.
Fatores de risco de craniossinostose.
- Baixo peso ao nascer
- Nascimento prematuro
- Idade paterna avançada
- Tabagismo materno durante a gravidez
Sintomas e Complicações
A maioria dos sintomas que podem ser observados na presença de craniossinostose é devida a um aumento na pressão dentro do crânio . Na medicina, o aumento da pressão dentro do crânio é chamado de hipertensão intracraniana ou hipertensão intracraniana .
Na presença de craniossinostose, a hipertensão intracraniana é uma consequência do fato de que o cérebro e outras estruturas dentro do crânio não têm o espaço certo para crescer, então eles vão empurrar as estruturas ósseas da cabeça.
Dito isso, é importante lembrar que, se as suturas cranianas envolvidas forem muitas ou se a condição não for tratada a tempo, a craniossinostose pode levar a um desenvolvimento reduzido de habilidades cognitivas e um baixo QI.
SINTOMAS DA HIPERTENSÃO ENDOCRÁGICA
Os possíveis sintomas de hipertensão intracraniana são:
- Dor de cabeça persistente. Geralmente piora de manhã e à noite.
- Problemas de visão. Consistem em visão dupla, visão turva e visão turva.
- vómitos
- irritabilidade
- Olhos inchados ou proeminentes
- Dificuldade em seguir o movimento dos objetos
- Problemas de audição
- Problemas respiratórios
- Alterações do estado mental
- papiledema
O número de suturas cranianas envolvidas no desenvolvimento da craniossinostose tem uma influência significativa na presença de hipertensão intracraniana.
Por exemplo, os médicos observaram que o envolvimento de apenas uma sutura craniana induz hipertensão intracraniana em 15% dos pacientes; enquanto o envolvimento de pelo menos duas suturas leva a um aumento na pressão dentro do crânio em pelo menos 60% dos pacientes.
Na presença de uma forma leve de craniossinostose, a hipertensão endocraniana começa a ser problemática, causando a sintomatologia supracitada, em torno de 4 a 8 anos de vida.
SINAIS DE CRANIOSINOSTOSE
Entre os sinais de craniossinostose, os mais comuns são:- Formações de cristas rígidas ao longo das suturas cranianas
- Anormalidades no nível da fontanela craniana
- Cabeça com dimensões não proporcionais ao resto do corpo
TIPOS DE CRANIOSINOSTOSE
A forma da cabeça dos pacientes com craniossinostose depende de quais suturas cranianas foram fechadas prematuramente.
Tendo observado isso, os médicos consideraram apropriado distinguir a craniossinostose em vários tipos, dependendo das suturas cranianas envolvidas.
Os tipos de craniossinostose são:
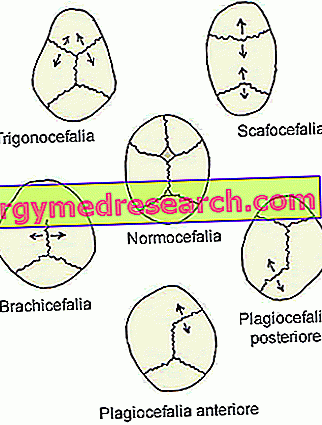
- Sinostose sagital ( dolichocefalia ou scafocephaly ). É o tipo mais comum de craniossinostose; na verdade, caracteriza cerca de metade dos casos clínicos.
Sua presença coincide com o fechamento prematuro das suturas cranianas sagitais, localizadas na parte superior do crânio, entre os ossos parietais.
De //en.wikipedia.org/wiki/Plagiocephaly
- Craniossinostose coronal ( braquicefalia ) É o segundo tipo de craniossinostose mais comum; Ele apresenta cerca de um caso clínico a cada quatro.
Seu início implica a fusão prematura das suturas coronais, que correm entre o osso frontal e os ossos parietais.
- Sinostose metópica ( trigonocefalia ). É um tipo bastante raro de craniossinostose, que distingue apenas 4-10% dos casos.
Sua aparência coincide com a fusão prematura da sutura metópica (ou frontal), que vai do nariz até o topo da cabeça, separando o osso frontal em dois. Normalmente, esta sutura naturalmente se ossifica no sexto ano de vida.
- Sinostose lambdoide ( plagiocefalia ). É o tipo mais raro de craniossinostose. De fato, só distingue 2-4% dos casos clínicos.
Sua presença envolve a fusão precoce da sutura lambdoide, localizada entre os ossos parietais e o osso occipital, na parte posterior da cabeça.

COMPLICAÇÕES
Além de afetar o desenvolvimento intelectual, uma craniossinostose não tratada pode determinar:
- A chamada síndrome da apneia obstrutiva do sono .
- Alterações faciais permanentes no nível dos olhos e ouvidos, em particular.
- Deformidades permanentes na base do crânio (por exemplo, a malformação ou síndrome de Arnold-Chiari).
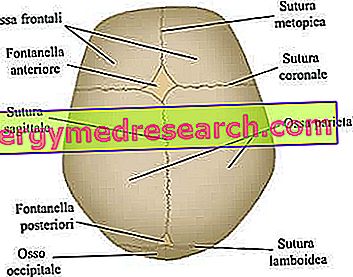
As principais suturas cranianas envolvidas no processo de craniossinostose. Do site: www.sciencebasedmedicine.org
- Hidrocefalia

diagnóstico
Para diagnosticar craniossinostose, exame objetivo, avaliação da história clínica e imagens radiológicas fornecidas por raios-X ou tomografia computadorizada na cabeça são essenciais.
Se a craniossinostose era do tipo sindrômico, também é importante estabelecer a condição mórbida que determinou seu início. Portanto, os médicos poderiam recorrer a exames de sangue e, sobretudo, a aconselhamento genético .
EXAME OBJETIVO
O exame objetivo consiste na análise cuidadosa, pelo médico, dos sinais clínicos presentes na cabeça do sujeito, suspeitos de sofrer de craniossinostose.
Geralmente, um pediatra está envolvido nesta importante verificação diagnóstica.
HISTÓRIA CLÍNICA
A avaliação da história clínica é importante para fins de diagnóstico, pois inclui questões relacionadas aos fatores de risco da craniossinostose.
Então, o médico (geralmente sempre um pediatra) irá investigar se:
- O bebê nasceu prematuro ou com baixo peso.
- Qual era a idade do pai no momento da concepção?
- Se a mãe fumou durante a gravidez.
EXAMES RADIOLÓGICOS
A radiografia e a tomografia computadorizada na cabeça servem mais do que qualquer outra coisa para confirmar o diagnóstico e mostrar, ao médico, quais suturas cranianas se fundiram prematuramente.
O conhecimento de quais suturas craniais estão envolvidas permite planejar o tratamento cirúrgico mais adequado.
tratamento
A craniossinostose é curável apenas por intervenção cirúrgica .
Este último consiste em uma operação de separação das suturas do fuselo craniano precocemente entre elas.
O objetivo terapêutico final da cirurgia é fornecer as estruturas cerebrais e alguns órgãos sensoriais, como os olhos, o espaço necessário para se desenvolver e funcionar da melhor maneira possível.
MELHOR MOMENTO PARA INTERVENIR
Não há acordo total por parte dos médicos sobre o melhor momento para realizar a cirurgia de craniossinostose.
Segundo alguns especialistas, o período ideal para a operação seria no final da infância, quando o risco de recaída (ou seja, uma segunda fusão prematura de suturas cranianas) é menor. Em caso de recidiva, de fato, a intervenção deve ser repetida e isso não é recomendado, dada a delicadeza do procedimento.
Segundo outros especialistas, o momento mais adequado seria na primeira infância (entre 6 e 12 meses de vida), quando o crânio ainda não está completamente ossificado e os ossos ainda estão modelando. A possibilidade de modelar os ossos (maleabilidade) permite resolver quaisquer anormalidades morfológicas dos próprios ossos, o que poderia causar sérios defeitos estéticos e problemas funcionais (na mandíbula ou nos olhos) em uma idade mais madura.
ABORDAGENS CIRÚRGICAS POSSÍVEIS
Existem duas abordagens cirúrgicas diferentes: cirurgia tradicional, também chamada de "ar livre", e cirurgia endoscópica.
- Cirurgia tradicional (ou "open air") .
Envolve anestesia geral (para que o paciente fique inconsciente durante todo o procedimento) e a prática de uma incisão cirúrgica na cabeça, exatamente onde as imagens radiológicas mostravam a anomalia craniana.
Através da incisão na cabeça, o cirurgião operacional (um neurocirurgião) remove o osso anormal e o confia a um especialista em cirurgia craniofacial, que o modifica e lhe dá uma forma que permite o desenvolvimento normal das estruturas encefálicas.
Após a modificação, o neurocirurgião substitui o osso em sua posição original e fecha a incisão com suturas.
Como muitas cirurgias tradicionais, a operação de "céu aberto" é bastante invasiva; entretanto, é vantajoso poder modificar a estrutura óssea de maneira precisa e com bons resultados.
- Cirurgia endoscópica .
Envolve o uso de um endoscópio, uma ferramenta semelhante a um tubo flexível, equipado com uma câmera de fibra óptica, em uma extremidade, e conectado a um monitor.
Do ponto de vista operatório, consiste em inserir o endoscópio em uma abertura feita no crânio e na separação, por meio do próprio endoscópio, da sutura fundida (sutura).
O neurocirurgião consegue se orientar dentro da cabeça, graças às imagens que a câmera projeta no monitor conectado externamente.
A cirurgia endoscópica é decididamente menos invasiva do que a operação de campo aberto (mesmo o período mais curto de hospitalização), no entanto, tem dois inconvenientes: só é indicado para pacientes de alguns meses (6 em geral), que possuem ossos que ainda podem ser modelados; está em maior risco de recaída.
FASE PÓS-OPERATÓRIA
Tipicamente, um paciente com craniossinostose, que foi submetido a cirurgia, deve permanecer no hospital por aproximadamente 4-5 dias após a operação. Durante esse tempo, o neurocirurgião e os membros de sua equipe monitoram periodicamente os parâmetros vitais e verificam se tudo corre bem.
Após a renúncia, prevê -se uma série de check-ups periódicos, que são, a princípio, semestrais e, depois, com o crescimento do paciente, anualmente.
prognóstico
O prognóstico depende de vários fatores, incluindo:
- Causas que causaram craniossinostose. Algumas doenças genéticas responsáveis por esta anomalia são muito graves e do mau prognóstico.
- A posição do fusel suturas prematuramente. Se as suturas residirem em posições que, para o neurocirurgião, são "inconvenientes" de se alcançar, a intervenção da craniossinostose se torna complicada e pode não fornecer os resultados desejados.


