generalidade
A doença de Huntington é uma doença devastadora, hereditária e neurodegenerativa, para a qual atualmente não há cura. De maneira lenta mas progressiva, a doença de Huntington reduz a capacidade de andar, falar e raciocinar. Finalmente, aqueles afetados pela doença de Huntington tornam-se completamente dependentes de outros para o tratamento.

Estima-se que 3 a 10 indivíduos sejam afetados por 100.000 indivíduos na Europa Ocidental e na América do Norte. Geralmente a idade de início varia entre 30 e 50 anos e a morte ocorre 15 a 20 anos após o início da doença. Também pode afetar crianças (juvenil Huntington); Neste caso, os indivíduos afetados raramente conseguem atingir a idade adulta.
A doença de Huntington afeta igualmente homens e mulheres e não faz distinção entre raças.
Os sintomas
Para saber mais: sintomas da doença de Huntington
Existem vários sintomas que afligem pacientes com doença de Huntington; os sintomas precoces podem envolver habilidades cognitivas ou motoras e incluem depressão, alterações de humor, esquecimento, falta de jeito, contrações involuntárias (córea) e falta de coordenação. Com a progressão da doença, a concentração e a memória de curto prazo diminuem, enquanto os movimentos da cabeça, do tronco e dos membros aumentam. A capacidade de andar, falar e engolir regride progressivamente, até que o indivíduo que sofre da doença de Huntington não seja mais capaz de cuidar de si mesmo. A morte geralmente ocorre como resultado de complicações como choque, infecção ou ataque cardíaco.
genética
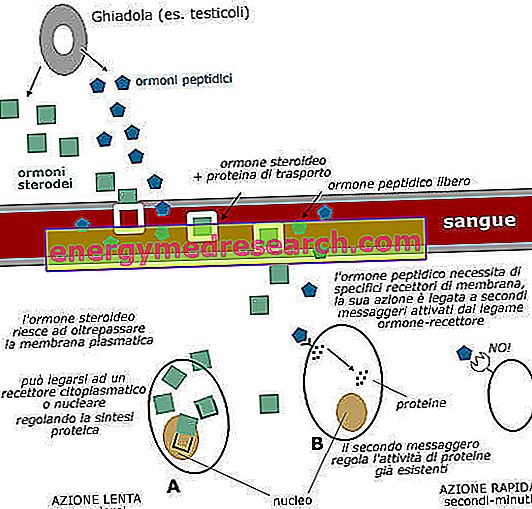
Em 1993 a mutação genética que causa a doença de Huntington foi descoberta, em um gene autossômico dominante, com penetrância incompleta mas muito alta, localizada no cromossomo 4. Este gene codifica uma proteína, chamada huntingtina ou HTT, cuja função ainda não é bem conhecida e é geralmente encontrada no citoplasma. Observou-se que a forma mutada de huntingtina contém uma secção de cadeia formada por resíduos de glutamina por muito mais tempo do que a presente na proteína normal. De facto, no gene não mutado, o codão que codifica a glutamina (CAG) é repetido 19-22 vezes, enquanto no gene mutado existe uma repetição até 48 vezes ou mais. Isto resultaria no alongamento dos resíduos de glutamina localizados na porção do terminal NH2 da proteína huntingtina.
Além disso, embora a proteína mutada seja expressa de forma ubíqua no corpo, a degeneração celular ocorre mais no cérebro. De fato, a doença de Huntington é caracterizada pela degeneração de neurônios do núcleo caudado, uma região dos gânglios (ou núcleos) da base responsável pela regulação do movimento voluntário.
Estudo aprofundado: gânglios da base, funções estriatais e neuropatologia da doença de Huntington
tratamento
Terapias farmacológicas têm um significado puramente sintomático e não influenciam a evolução da doença ou seu processo degenerativo. Por exemplo, os antagonistas da dopamina podem ser usados para aliviar os movimentos coreicos. No entanto, seu uso é limitado para efeitos indesejáveis, como sedação e depressão. As drogas antiparkinsonianas, por outro lado, podem influenciar positivamente as formas jovens dominadas pela rigidez. Os transtornos psicóticos podem requerer tratamento psicofarmacológico adequado (neurolépticos, sais de lítio), enquanto os sintomas depressivos podem ser atenuados pelo uso de drogas específicas (antidepressivos tricíclicos, serotoninérgicos).
Apesar de numerosos ensaios clínicos conduzidos na última década, nenhuma droga foi demonstrada até o momento em um estudo randomizado com placebo na terapia da doença de Huntington. A fase clínica é muito exigente, principalmente porque a doença tem uma progressão lenta e uma ampla heterogeneidade clínica. As escalas de avaliação da doença de Huntington existem e são praticamente as mesmas em todas as clínicas. A penetrância completa da doença e a disponibilidade de testes genéticos preditivos oferecem a oportunidade de tentar o tratamento durante os estágios iniciais da doença. Atualmente os estudos são voltados para a pesquisa de biormarcadores de mudanças, sensíveis e estáveis, a fim de intervir nas primeiras manifestações da doença.
Atualmente, as técnicas de neuroimagem oferecem os melhores biomarcadores durante a fase prodrômica (que precede os sintomas clínicos da doença); Eles também fornecem uma correlação entre as terapias realizadas em modelos animais e em humanos. Como mencionado, a atrofia do estriado é precoce e progride durante o curso da doença. Também foi demonstrado que outras áreas do cérebro, como as estruturas subcorticais e corticais da substância branca, são afetadas no período prodrômico.
Através de imagens funcionais também pode identificar algumas anormalidades em indivíduos durante o período prodrômico. Esta técnica pode revelar-se sensível o suficiente para identificar irregularidades na estrutura detectável ou mudanças comportamentais.
Finalmente, a identificação de biomarcadores moleculares, como o lactato ou outros produtos de estresse celular, poderia ser permitida graças às técnicas de espectroscopia de ressonância magnética.
Doença de Huntington e Receptores Canabinoides »